
Maladies métaboliques
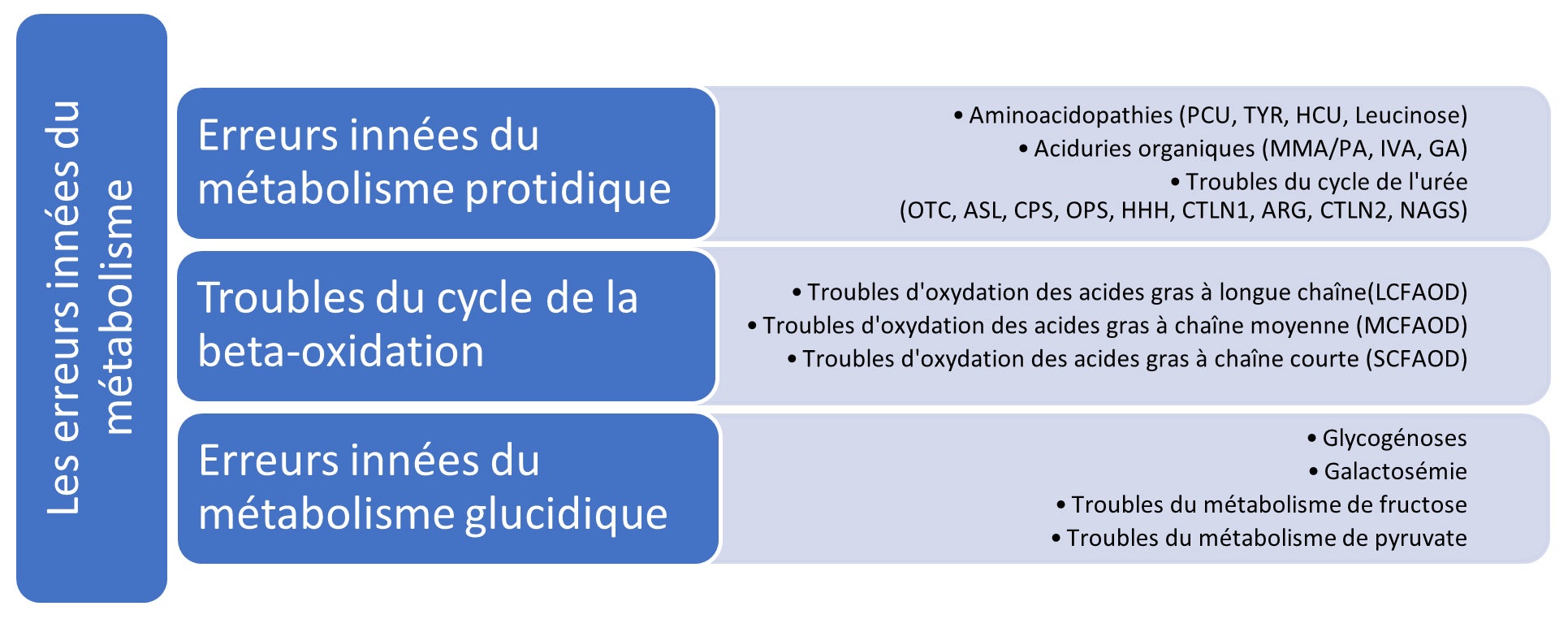
Les erreurs innées du métabolisme sont des troubles génétiques héréditaires dans lesquels un défaut enzymatique spécifique perturbe le métabolisme normal des protéines, des lipides ou des glucides. En raison de l’activité enzymatique réduite ou absente dans ces troubles, des composés spécifiques s’accumulent à des niveaux toxiques dans le corps. Cela peut également empêcher le corps de produire de manière adéquate les composés spécifiques dont il a besoin, entraînant des carences. Ces perturbantes métaboliques, si elles ne sont pas traitées, peuvent entraîner une série de résultats médicaux et développementaux, allant de troubles cognitifs, de défaillances d’organes et même de décès.
Beaucoup de conséquences négatives d’erreurs innées du métabolisme peuvent être atténuées par une détection et une prise en charge précoces qui peuvent inclure une intervention médicamenteuse et/ou diététique, en utilisant des denrées alimentaires destinées à des fins médicales spéciales (DADFMS).
Individuellement, les erreurs innées du métabolisme sont rares mais collectivement plus fréquentes (3,5 – 5,9 % de la population mondiale (1)). De nombreuses maladies rares héréditaires sont dépistées à la naissance via le test de piqûre au talon, ce dépistage néonatal permet une identification précoce et l’initiation d’une prise en charge améliorant les résultats pour les patients. L’efficacité du dépistage néonatal a entraîné une augmentation des taux d’incidence des erreurs innées du métabolisme et bien que le dépistage soit recommandé, tous les pays ne l’ont pas mis en place.
La Phénylcétonurie (PCU) est l’une des erreurs innées du métabolisme les plus courantes qui nécessite une prise en charge nutritionnelle, et a un taux d’incidence global d’environ 1 sur 10.000 à 1 sur 12.000 (2).
La PCU est une maladie héréditaire qui nécessite que le gène PCU soit hérédité des deux parents et n’est pas liée au sexe, ce qu’on appelle autosomique récessif. La PCU est un trouble du métabolisme des protéines, spécifiquement lié à l’acide aminé: phénylalanine (les acides aminés sont les éléments constitutifs des protéines). La PCU est caractérisée par un déficit en enzyme hépatique connue sous le nom de phénylalanine hydroxylase qui convertit la phénylalanine (Phé) en tyrosine (Tyr), entraînant une accumulation de Phé et un déficit en Tyr dans le sang.
En l’absence d’une prise en charge, cela peut entrainer des niveaux toxiques de Phé, entraînant des lésions cérébrales irréversibles entraînant des difficultés d’apprentissage, des changements de comportement, des tremblements et convulsions. Des faibles taux de Tyr entraînent une altération de la production de neurotransmetteurs et une carence en mélanine, provoquant un teint pâle caractéristique. Les taux de Phé et de Tyr doivent être surveillés dans la PCU en prélevant régulièrement des gouttes de sang et en gérant l’apport en protéines naturelles.
La Phé étant un acide aminé essentiel, il est nécessaire au fonctionnement normal du corps et ne peut pas être produit par le corps. Pour éviter que la Phé s’accumule à des taux toxiques, une petite quantité bien mesurée de protéines naturelles doit encore être consommée pour s’assurer que les besoins de Phé sont satisfaits mais pas dépassés. Les personnes atteintes de PCU ont différentes tolérances à la Phé en fonction de la gravité de leur état, de leur âge et de leur croissance.
Les protéines sont essentielles à la croissance et à la réparation. Les petites quantités de protéines naturelles consommées par les patients atteints de PCU ne suffisent pas à couvrir les besoins totaux en protéines. Des produits appelés des substitutes de protéines (PS) sont prescrits afin d’atteindre les niveaux de protéines requis par les patients.
Divers termes sont utilisés pour décrire les substituts de protéines, notamment les préparations spéciales et les mélanges d’acides aminés. Les substituts de protéines contiennent ce qu’on appelle un “équivalent protidique », ce qui signifie que ces produits contiennent tous les acides aminés présents dans des protéines, mais avec l’acide aminé nocif, dans ce cas la Phé, entièrement ou fortement réduit. Des nutriments supplémentaires sont ajoutés aux PS en raison de la nature restrictive du régime alimentaire afin d’éviter des carences nutritionnelles. Il existe deux types principaux de substituts de protéines disponibles pour la PCU – les PS à base d’acides aminés et les PS à base de glycomacropeptide (GMP). Des aliments hypoprotidiques manufacturés sont également disponibles pour l’utilisation dans la gestion diététique de la PCU, ainsi que pour d’autres troubles métaboliques. Il s’agit notamment d’aliments de base tels que du pain hypoprotidique, des pâtes et des alternatives au lait permettant plus de normalité dans le régime alimentaire tout en fournissant de l’énergie supplémentaire pour soutenir le contrôle métabolique global.
Un diagnostic précoce et une intervention diététique peuvent améliorer considérablement les résultats pour les patients (3).
Plus d’informations sur les erreurs innées du métabolisme et la prise en charge diététique peuvent être trouvées sur Vitaflo | Nestlé Health Science (nestlehealthscience.com)
Toute personne affectée par une erreur innée du métabolisme souhaitant plus d’informations sur la maladie, la prise en charge et les résultats, doit demander l’avis médical de son médecin et/ou son diététicien.
Références:
- Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. European Journal of Human Genetics 2019 28:2 [Internet]. 2019 Sep 16 [cited 2021 Oct 21];28(2):165–73. Available from: https://www.nature.com/articles/s41431-019-0508-0
- Dixon M, MacDonald A, White F, Stafford J. Disorders of Amino Acid Metabolism, Organic Acidaemias and Urea Cycle Disorders. Clinical Paediatric Dietetics: Fourth Edition [Internet]. 2014 Nov 17 [cited 2021 Oct 13];381–525. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/9781118915349.ch17
- Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Archives of Disease in Childhood [Internet]. 2007 Mar 1 [cited 2021 Oct 15];92(3):213–8. Available from: https://adc.bmj.com/content/92/3/213
DÉCOUVREZ ÉGALEMENT
